



什么是液相色谱?
液相色谱(liquid chromatography,LC)是一种色谱技术,用于分离和分析溶液中混合物的化学成分,以确定是否存在或不存在特定成分,如果存在,则存在多少。我们中的许多人会从上学开始就熟悉平面LC的形式,在滤纸上打上黑色墨水标记,将一端浸入水中,然后观察墨水中的成分颜色是否分开。但是,分析应用中使用的大多数LC均基于柱色谱法,这将是本文的重点。顾名思义,高效液相色谱(High Performance Liquid Chromatography,HPLC)是使用高色谱分辨率进行高效分离的高性能分析。分离的组分也可以在检测后使用馏分收集器分离,作为纯化的手段。HPLC有多种不同的配置,可用于分离分子量从半挥发性小分子到几万千道尔顿的大蛋白生物分子的溶解组分。液相色谱法是一种非常流行的分析技术,广泛用于环境监控,农业,医药领域。
液相色谱如何工作?
液相色谱仪有多种不同的系统配置,其中最高效率的分离是在超高效液相色谱(UHPLC)仪器上进行的,该技术于2004年首次商业化,被称为超高效液相色谱(UPLC)。简而言之,由于各个组分在流动相(下图1(1))和固定相(柱)(图1(3))之间的独特分配,可溶于液相流动相的多组分混合物得以分离。
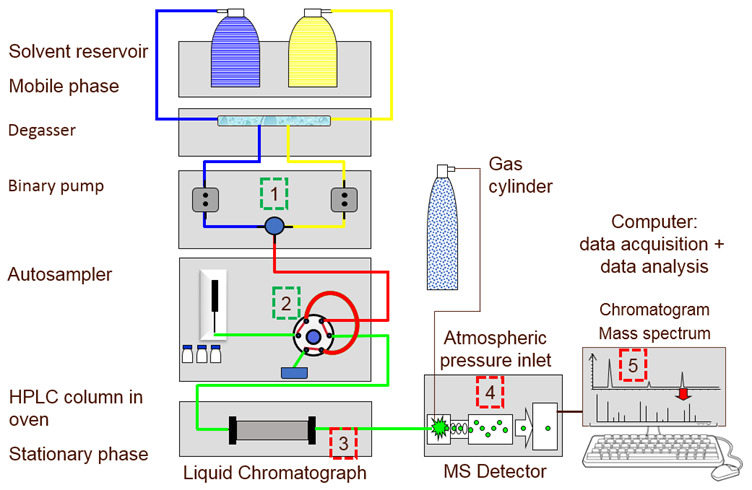 图1:连接质谱仪(LC-MS)的液相色谱仪简化图。(1)用于流动相的二元泵,(2)自动进样器的6通阀和进样器环路,(3)色谱柱加热器(4)质谱仪检测器,(5)PC。
图1:连接质谱仪(LC-MS)的液相色谱仪简化图。(1)用于流动相的二元泵,(2)自动进样器的6通阀和进样器环路,(3)色谱柱加热器(4)质谱仪检测器,(5)PC。
流动相通常是溶剂,借助高压泵(图1(1))将样品输送并且通过系统。它在分离过程中也起着至关重要的作用。
将少量样品(1-100 µL)装入样品定量环(图1(2)),然后通过六通阀注入流动相中,这会触发色谱的启动。进样后,将流动相泵入色谱柱(图1(3))。有多种色谱柱长度(30到250毫米)和内径(1到4.6毫米)可供选择,填充有不同活性和粒径(1.5到10微米直径)的固定相吸附材料,共同决定了色谱柱的效率和选择性。该柱位于柱箱中。在较高温度(45ºC)下,流动相的粘度降低,从而增加了其线速度。反过来,减少了运行时间,也提高了色谱分离度。
混合物中对流动相具有较高亲和力的组分将快速迁移通过色谱柱,而与固定相之间几乎没有相互作用。当组分的带离开色谱柱或从色谱柱洗脱时,检测器(图1(4))将给出与组分浓度成正比的响应。进样与检测之间的时间称为保留时间。对于一组给定的色谱条件,组分的保留时间将非常明确,可以与鉴定标准进行比较。
在反相色谱法中,极性较小的分析物会优先分配到非极性固定相中,并且保留时间更长。数据采集系统(图1(5))将检测器响应记录为色谱图中保留时间的函数。通常对色谱图中记录的峰(下图2)进行积分以确定峰面积,该峰面积与样品中存在的组分的浓度成正比。
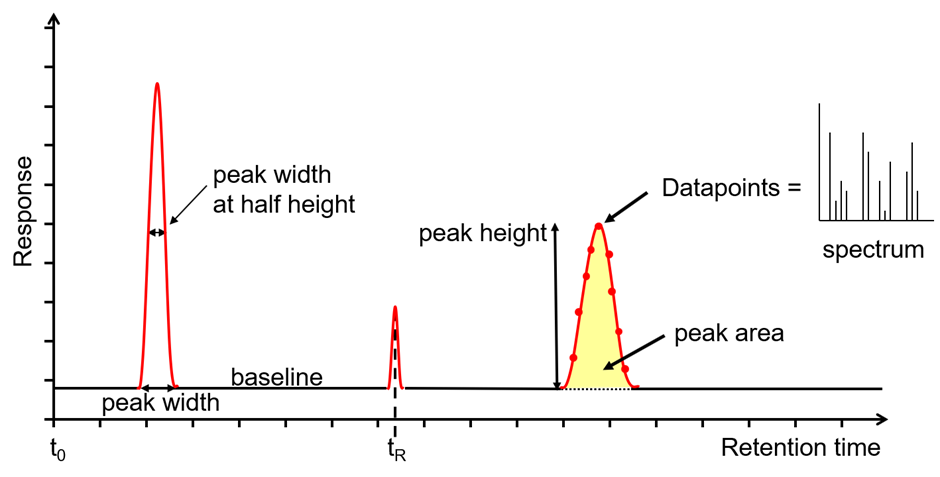
图2:HPLC或LC-MS的色谱图输出。
自动进样器将样品注入流动相后,在色谱柱中进行分离过程。色谱系统的选择性对色谱分辨率的影响最大,应针对所研究的应用和组件进行定制。选择性可以通过改变流动相(不同溶剂)或固定相中存在的特定化学官能团(改变色谱柱类型)的电化学强度来改变。
考虑到流动相,运行液相色谱仪时有两种主要的操作模式可供选择,即等度或梯度。等度方法将在色谱运行期间使用相同的流动相组成,而选择性没有变化。梯度方法将使流动相的组成随时间变化,通常可以对其进行优化以提高色谱分辨率或缩短运行时间。
固定在色谱柱内的固定相的化学性质会影响该技术的选择性。反相HPLC或UHPLC是最流行的系统配置,并使用非极性固定相(例如十八烷基硅烷(ODS或C18))和极性流动相(水/甲醇)。其他反相固定相包括八硅烷(C8),它的疏水性比C18小,对极性较小的分析物的保留时间也相应较短。如果使用被酚取代基官能化的色谱柱,由于其亲和力的提高,这将增加酚组分的保留。
流动相的pH对离子组分的保留时间有深远的影响,在方法开发过程中应加以利用。缓冲液可用于将流动相的pH维持在离子组分的pK a以下两个单位,这反过来将其解离平衡转变为中性形式。组件的中性形式极性较小,因此可以控制其保留时间。
正相色谱法是另一种液相色谱方法,可根据其极性分离分析物,实际上是在引入反相液相色谱法之前开发的,但不太普及。固定相在正相色谱法中为极性流动相是非极性的。这改变了系统的保留特性,混合物的非极性组分首先以最短的保留时间被洗脱。极性分析物对固定相的亲和力更高,随后洗脱时间更长,保留时间更长。还有其他类型的液相色谱,包括离子色谱,离子对,尺寸排阻,亲和力,列表不胜枚举。除了尺寸排阻色谱法可以根据其大小/形状或分子量分离分析物外,上述提及的其他形式的液相色谱仪均采用不同的流动相和固定相化学方法。对于给定的一组要分离的组分,可获得的选择性和色谱分离度由所用的固定相和流动相定义。
组件一旦分离,就需要进行检测。探测器的选择由应用程序的方法目标驱动;各种选项都具有不同程度的灵敏度,特异性,选择性和线性动态范围。最受欢迎的检测器是紫外线可见(UV-Vis)检测器,该检测器可测量特定波长下光的吸收率。根据要分析的组分的λ最大值选择波长,检测器的响应与该特定组分的浓度成正比。随着组分从色谱柱上洗脱下来,其在检测器流通池中的浓度会上升和下降,然后将其绘制为色谱峰(参见图2)。数据采集速率应设置为在整个峰中至少采集20个数据点。与许多色谱技术一样,
用于互连LC系统各个组件的管道的长度和内径至关重要,应保持绝对最小。从注入回路开始到检测器流通池末端的色谱系统的任何部分(不是固定相)都无助于有效分离。系统内的这种额外体积称为空隙体积,空隙中分离成分的额外纵向扩散将导致灵敏度损失和色谱分离度降低。
HPLC到UHPLC从的演变
UHPLC的发展部分是由于分析人员对日益复杂和具有挑战性的样品进行更高分辨率分离的要求不断提高。使色谱性能发生这一阶段变化的主要突破是开发了具有窄粒度分布的亚2微米固定相填料。
生产的新颗粒具有与常用HPLC固定相相同的化学功能,可确保在使用相同的流动相时保持色谱系统的选择性。当使用新的亚2微米包装材料时,效率的提高或板数的增加实现了显着的性能优势。
将HPLC方法迁移到UHPLC系统时,有许多优点,包括运行时间更短,色谱分离度更高,灵敏度更高以及溶剂消耗更少。为了使用新型UHPLC色谱柱,必须使用可以在较高压力下运行的泵,以适应色谱柱中较小颗粒施加的增加的背压。检测器流通池还需要升级以具有较小的内部体积,这对于检测从色谱柱上洗脱下来的组分的较窄谱带是必需的。还需要相应地提高数据采集速率,以确保峰值之间有足够的数据点。
液质联用(LC-MS)
质谱分析可以说是最好的可与液相色谱仪联用的检测器,因为它在使用具有很高质量分辨能力的仪器时具有很高的灵敏度,线性动态范围,选择性甚至特异性。质谱技术用于确定组分或分析物的质荷比(m/z)。与气相色谱-质谱法(GC-MS)不同,液相色谱系统向质谱联用的过程并不容易,并且需要花费很多年的开发时间。电喷雾电离(ESI)是当今LC-MS中最常用的电离技术,其中电离过程在大气压下进行。很难达到质谱系统所需的高真空的大气压入口。
如何读懂LC-MS质谱图,它告诉您什么?
电喷雾电离是一种非常柔软的电离技术,这意味着在离子形成过程中观察到的碎片很少。对于生成质子化分子[M+H] +的碱性分析物,可以在正离子模式下运行,对于生成去质子化分子[MH] -的酸性分析物,可以在负离子模式下运行LC-MS系统。
是可能的碎片离子通过电喷射过程通常是通过碰撞诱导解离(CID),以获得用于表征或靶分析物的识别信息生成的。CID可以在离子源中通过更改施加到第一采样锥或撇渣锥的电势差来执行,也可以在碰撞池中将离子加速到诸如氩气的碰撞气体中执行。
下图3表示全扫描LC-MS采集,其中源内碰撞引起解离,从而为混合物的每个分离组分产生一系列特征性碎片离子。质荷比(m/z)沿x轴绘制,离子的强度或相对丰度沿y轴绘制。图3上的z轴代表分离的组分的保留时间,每个基线解析色谱峰一次用质谱仪分析一次,关键的诊断碎片离子可用于鉴定和目标离子确认。

图3: 全扫描LC-MS质谱图,MS添加了额外的信息维度。
将液相色谱法纳入多个维度
当处理复杂的多组分混合物时,可能无法基线解析每个组分,因为各个峰会从色谱柱上洗脱下来。如果由于缺乏选择性而无法提供色谱分离度,那么方法开发和优化只能帮助您解决问题,那么可能有必要使用其他具有选择性的色谱柱来分离共洗脱组分。
多维色谱可使共洗脱组分通过“心切”法以更合适的选择性转移到另一个色谱柱上,从而可以将分离拆分为单个洗脱组分。该系统可以设置有转移阀,该转移阀将检测后的第一维或色谱柱中的共洗脱峰简单地转移到第二维中,以进行后续分离和更高的色谱分离度检测。
半制备液相色谱法纯化
通过扩大液相色谱系统的尺寸,使用具有较大内径且以较高流速运行的色谱柱,可以将更多的材料装载到色谱柱上。半制备色谱系统可能装有100毫克的样品。然后使用馏分收集器将色谱峰从色谱柱上洗脱下来,收集到单独的样品瓶中。馏分收集器由检测器触发,检测器在色谱基线中寻找表明峰开始的拐点,然后将分离出的组分的峰收集为纯馏分。然后可以对分离出的馏分进行补充质谱的其他分析技术,例如核磁共振(NMR),以充分表征化合物的结构说明。
液相色谱法的优缺点
LC通常用于各种应用。但是,它不适用于挥发性化合物的分离和分析。仅当所有要分离的组分的蒸气压低于流动相的蒸气压时,才能实现可靠的分析型液相色谱方法。气相色谱法更适合分析挥发性化合物。
提供各种不同的色谱柱和溶剂,可提供广泛的选择性,从而可以分离极性范围很广的组分。大分子和小分子同样适用于该技术。在相对较低的温度下进行有效分离的能力也使LC成为可在气相色谱仪中分解的热不稳定化合物的理想分离技术。
液相色谱法的常见问题
样品准备是成功的关键;在将所有样品加载到自动进样器中之前,必须对所有样品进行过滤,这一点非常重要。当使用UHPLC时,如果不过滤样品,使用亚2微米颗粒的色谱柱进行高效分离时很容易发生阻塞,这一点就尤为重要。对于流动相也是如此,尤其是在使用缓冲区的情况下。
对于样品使用正确的进样溶剂或稀释剂至关重要,溶剂强度应等于或小于流动相起始条件的强度。如果使用太强的溶剂,则会观察到峰分裂和重现性差。如果自动进样器中使用的清洗溶剂太强,可能会观察到类似的问题。
所获取色谱图的基线波动或保留时间的重现性差,很可能是由于泵(图1(1))或真空脱气机出现问题而导致的。如果泵或真空脱气机维护不当,止回阀可能会部分卡住,从而引起压力波动。这些问题可以通过确保按照制造商指南执行预防性维护任务来解决,以防止计划外的停机时间和较差的性能。
参考文章:
1. García‐Alvarez‐Coque MC, Baeza‐Baeza JJ, Ramis‐Ramos G. Reversed Phase Liquid Chromatography. In: Analytical Separation Science. American Cancer Society; 2015:159-198. doi:10.1002/9783527678129.assep008
2. Zhao JJ, Yang AY, Rogers JD. Effects of liquid chromatography mobile phase buffer contents on the ionization and fragmentation of analytes in liquid chromatographic/ionspray tandem mass spectrometric determination. J Mass Spectrom. 2002;37(4):421-433. doi:10.1002/jms.299
3. Cooper WT. Normal-Phase Liquid Chromatography. In: Encyclopedia of Analytical Chemistry. American Cancer Society; 2006. doi:10.1002/9780470027318.a5913
4. Ibrahim D, Ghanem A. Sub-2 μm Silica Particles in Chiral Separation. New Uses of Micro and Nanomaterials, IntechOpen. doi: 10.5772/intechopen.79063
5. Stoll DR, Carr PW. Two-Dimensional Liquid Chromatography: A State of the Art Tutorial. Anal Chem. 2017;89(1):519-531. doi:10.1021/acs.analchem.6b03506
6. Miliauskas G, van Beek TA, de Waard P, Venskutonis RP, Sudhölter EJR. Comparison of analytical and semi-preparative columns for high-performance liquid chromatography-solid-phase extraction-nuclear magnetic resonance. J Chromatogr A. 2006;1112(1-2):276-284. doi:10.1016/j.chroma.2005.11.059





![[转载]高熵合金催化剂浅谈-如意](https://rueee.com/wp-content/uploads/2022/12/165509keqm3ltm3vmvayie-300x300.png)





暂无评论内容